Context of genetic population clustering
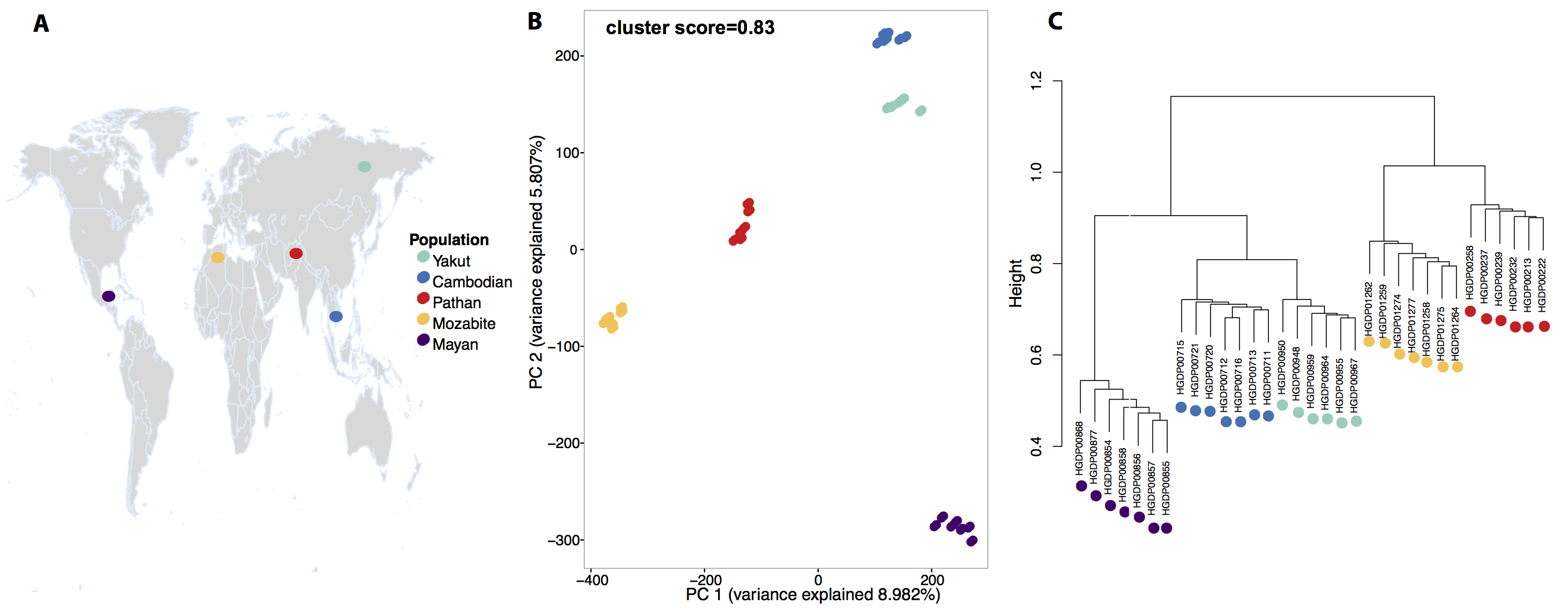
To characterize genetic divergence between these populations, we carried out PCA on the SNP genotype matrix. The first and second principal components clearly differentiated the individuals into five well-separated clusters that correspond to the five populations sampled. Even with the limited sample size, the population structure revealed by the SNP genotypes is extremely robust. To facilitate comparison between the genetic and epigenetic datasets we quantified the strength of the genomic PCA clustering by computing the Silhouette cluster scores (SCS) for the individuals in the five populations as well as the average SCS for the entire data set (Supplementary Figure S1). The SCS of an individual measures how similar it is to its own predefined population cluster, relative to individuals in other clusters, while the average SCS across all individuals is a measure of how tightly the data correspond to their known populations. For the genetic clustering presented in Figure 1B this score is 0.83, and the median is 0.9. A tree generated using hierarchical clustering also captures the genetic relationships between the individuals and their populations (Figure 1C). The branching pattern of this tree agrees with the accepted order of ancestral human expansion, consistent with the “out of Africa” hypothesis.