|
H. Takenaka, I. Grinberg, and A. M. Rappe, "Anisotropic local correlations and dynamics in a relaxor ferroelectric", Phys. Rev. Lett. 110, 147602 (1-5) (2013).PDF
|
|
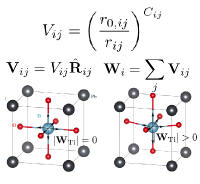
S. Liu, I. Grinberg, and A. M. Rappe, "Development of a bond-valence based interatomic potential for BiFeO3 for accurate molecular dynamics simulations", J. Phys. Cond. Matt. 25, 102202 (1-6) (2013).
PDF
|
|
L. Jiang, S. V. Levchenko, and A. M. Rappe,
"Rigorous Definition of Oxidation States of Ions in Solids",
Phys. Rev. Lett. 108, 166403 (1-5) (2012).
PDF
Supplementary material
PDF
|
|
T. Qi, Y.-H. Shin, K.-L. Yeh, K. A. Nelson, and A. M. Rappe,
"Collective Coherent Control: Synchronization of Polarization in
Ferroelectric PbTiO3 by Shaped THz Fields", Phys. Rev. Lett.
102, 247603 (1-4) (2009).
PDF |
|
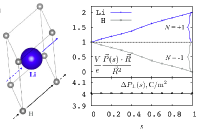
Y.-H. Shin, B.-J. Lee, and A. M. Rappe,
"Bond-valence model of Ferroelectric PbTiO3", J. Kor. Phys.
Soc. 52, 1206-10 (2008).
PDF
|
|
Y.-H. Shin, J.-Y. Son, B.-J. Lee, I. Grinberg, and
A. M. Rappe, "Order-disorder character of
PbTiO3", J. Phys.: Cond. Matt. 20, 015224 (1-5) (2008).
PDF
|
|
Y.-H. Shin, I. Grinberg, I.-W. Chen, and A. M. Rappe,
"Nucleation and growth mechanism of ferroelectric domain-wall motion",
Nature 449, 881-6 (2007).
PDF
Supplementary material
PDF
|
|
Y.-H. Shin, V. R. Cooper, I. Grinberg, and A. M. Rappe,
"Development of a bond-valence molecular-dynamics model for complex
oxides", Phys. Rev. B 71, 054104 (1-4) (2005).
PDF
|